Introduction to HiCExperiment
Jacques Serizay
2026-04-29
Source:vignettes/HiCExperiment.Rmd
HiCExperiment.RmdIntroduction
Hi-C experimental approach allows one to query contact frequency for all possible pairs of genomic loci simultaneously, in a genome-wide manner. The output of this next-generation sequencing-supported technique is a file describing every pair (a.k.a contact, or interaction) between two genomic loci. This so-called “pairs” file can be binned and transformed into a numerical matrix. In such matrix, each cell contains the raw or normalized interaction frequency between a pair of genomic loci (which location can be retrieved using the corresponding column and row indices).
HiC-Pro, distiller and Juicer are the three main pipelines used to align, filter and process paired-end fastq reads into pairs files and contact matrices. Each pipeline defined their own file formats to store these two types of files.
Pairs files are (gzipped) human-readable, text files that are a variant of the BEDPE format; however the column order varies depending on the pipeline being used.
-
Contact matrix file formats greatly vary depending on the pipeline:
-
HiC-Progenerates two human-readable files: aregionsfile describing each genomic interval, and amatrixfile quantifying interaction frequency between pairs of loci from theregionsfile, using a standard triplet sparse matrix format. -
Juicergenerates a.hicfile, a highly compressed binary file storing sparse contact matrices from multiple resolutions into a single file. -
distilleruses the.(m)coolformat, a sparse, compressed, binary genomic matrix data model built on HDF5.
-
Each file format can contain roughly the same information, albeit
with a largely improved compression for .hic and
.(m)cool files, which can also contain multi-resolution
matrices compared to the HiC-Pro derived files. The 4DN
consortium, deciphering the role nuclear organization plays in gene
expression and cellular function, officially supports both the
.hic and .(m)cool formats. Furthermore, the
.(m)cool format has recently gained a lot of traction with
the release of a series of python packages
(cooler, cooltools, pairtools,
coolpuppy) by the Open2C organization facilitating
the investigation of Hi-C data stored in .(m)cool files in
a python environment.
The R HiCExperiment package aims at unlocking HiC
investigation within the rich, genomic-oriented Bioconductor
environment. It provides a set of classes and import functions to parse
HiC files (both contact matrices and pairs) in R, allowing random access
and efficient genome-based subsetting of contact matrices. It leverages
pre-existing base Bioconductor classes, notably
GInteractions and ContactMatrix classes (Lun, Perry &
Ing-Simmons, F1000 Research 2016).
Installation
HiCExperiment package can be installed from Bioconductor
using the following command:
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("HiCExperiment")All R dependencies will be installed automatically.
The HiCExperiment class
library(HiCExperiment)
showClass("HiCExperiment")
#> Class "HiCExperiment" [package "HiCExperiment"]
#>
#> Slots:
#>
#> Name: fileName focus resolutions
#> Class: character characterOrNULL numeric
#>
#> Name: resolution interactions scores
#> Class: numeric GInteractions SimpleList
#>
#> Name: topologicalFeatures pairsFile metadata
#> Class: SimpleList characterOrNULL list
#>
#> Extends: "Annotated"
#>
#> Known Subclasses: "AggrHiCExperiment"
hic <- contacts_yeast()
#> see ?HiContactsData and browseVignettes('HiContactsData') for documentation
#> loading from cache
hic
#> `HiCExperiment` object with 8,757,906 contacts over 763 regions
#> -------
#> fileName: "/github/home/.cache/R/ExperimentHub/3e261f048867_7752"
#> focus: "whole genome"
#> resolutions(5): 1000 2000 4000 8000 16000
#> active resolution: 16000
#> interactions: 267709
#> scores(2): count balanced
#> topologicalFeatures: compartments(0) borders(0) loops(0) viewpoints(0) centromeres(16)
#> pairsFile: N/A
#> metadata(0):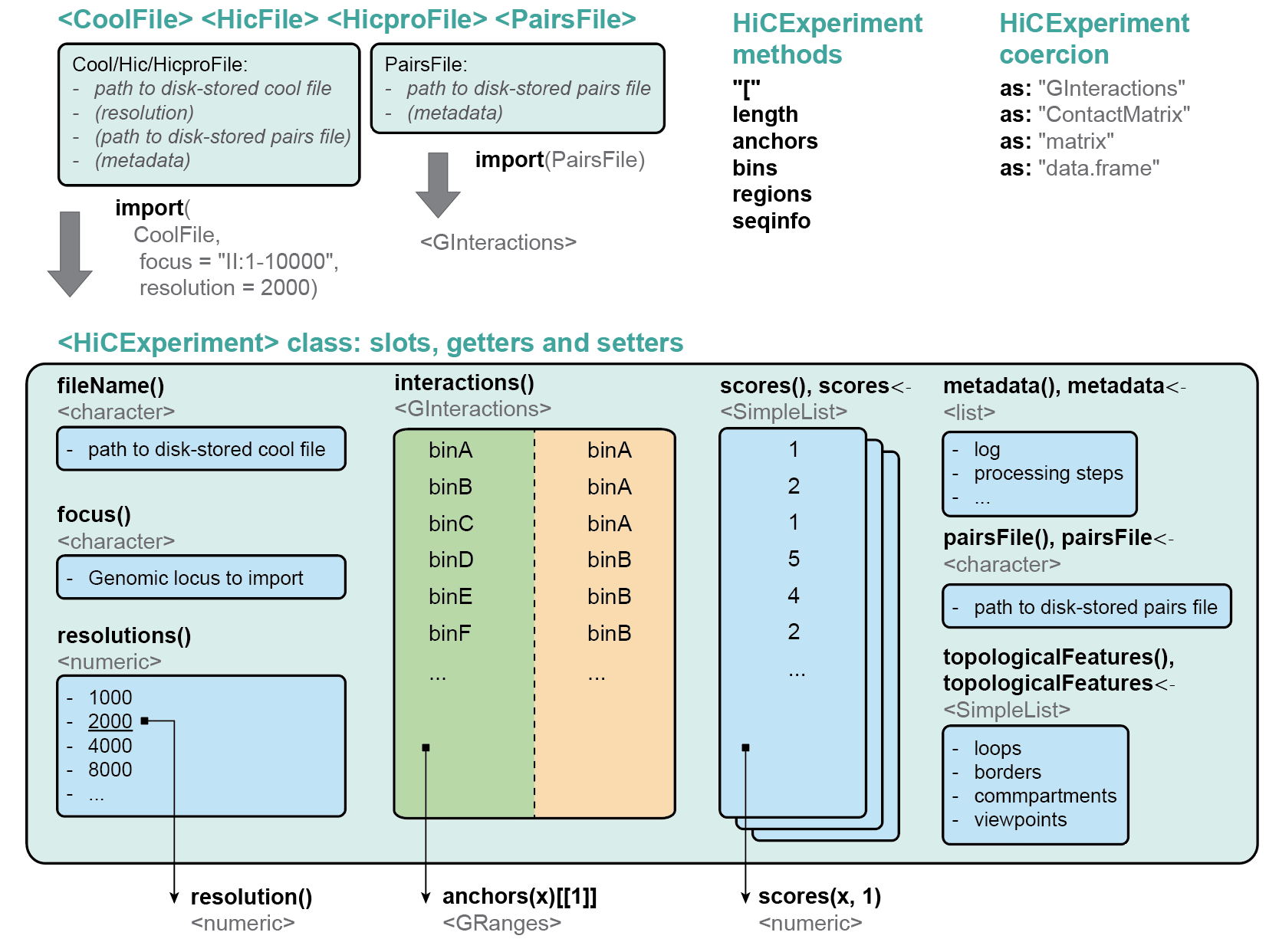
Basics: importing .(m)cool, .hic or
HiC-Pro-generated files as HiCExperiment objects
Import methods
The implemented import() methods allow one to import
Hi-C matrix files in R as HiCExperiment objects.
## Change <path/to/contact_matrix>.cool accordingly
hic <- import(
"<path/to/contact_matrix>.cool",
focus = "chr:start-end",
resolution = ...
)To give real-life examples, we use the HiContactsData
package to get access to a range of toy datasets available from the
ExperimentHub.
library(HiContactsData)
cool_file <- HiContactsData('yeast_wt', format = 'cool')
#> see ?HiContactsData and browseVignettes('HiContactsData') for documentation
#> loading from cache
import(cool_file, format = 'cool')
#> `HiCExperiment` object with 8,757,906 contacts over 12,079 regions
#> -------
#> fileName: "/github/home/.cache/R/ExperimentHub/3e2645368057_7751"
#> focus: "whole genome"
#> resolutions(1): 1000
#> active resolution: 1000
#> interactions: 2945692
#> scores(2): count balanced
#> topologicalFeatures: compartments(0) borders(0) loops(0) viewpoints(0)
#> pairsFile: N/A
#> metadata(0):Supporting file classes
There are currently three main standards to store Hi-C matrices in files:
-
.(m)coolfiles -
.hicfiles -
.matrixand.bedfiles: generated by HiC-Pro.
Three supporting classes were specifically created to ensure that
each of these file structures would be properly parsed into
HiCExperiment objects:
CoolFileHicFileHicproFile
For each object, an optional pairsFile can be associated
and linked to the contact matrix file when imported as a
HiCExperiment object.
## --- CoolFile
pairs_file <- HiContactsData('yeast_wt', format = 'pairs.gz')
#> see ?HiContactsData and browseVignettes('HiContactsData') for documentation
#> loading from cache
coolf <- CoolFile(cool_file, pairsFile = pairs_file)
coolf
#> CoolFile object
#> .mcool file: /github/home/.cache/R/ExperimentHub/3e2645368057_7751
#> resolution: 1000
#> pairs file: /github/home/.cache/R/ExperimentHub/3e2648419160_7753
#> metadata(0):
import(coolf)
#> `HiCExperiment` object with 8,757,906 contacts over 12,079 regions
#> -------
#> fileName: "/github/home/.cache/R/ExperimentHub/3e2645368057_7751"
#> focus: "whole genome"
#> resolutions(1): 1000
#> active resolution: 1000
#> interactions: 2945692
#> scores(2): count balanced
#> topologicalFeatures: compartments(0) borders(0) loops(0) viewpoints(0)
#> pairsFile: /github/home/.cache/R/ExperimentHub/3e2648419160_7753
#> metadata(0):
import(pairsFile(coolf), format = 'pairs')
#> GInteractions object with 471364 interactions and 3 metadata columns:
#> seqnames1 ranges1 seqnames2 ranges2 | frag1 frag2
#> <Rle> <IRanges> <Rle> <IRanges> | <numeric> <numeric>
#> [1] II 105 --- II 48548 | 1358 1681
#> [2] II 113 --- II 45003 | 1358 1658
#> [3] II 119 --- II 687251 | 1358 5550
#> [4] II 160 --- II 26124 | 1358 1510
#> [5] II 169 --- II 39052 | 1358 1613
#> ... ... ... ... ... ... . ... ...
#> [471360] II 808605 --- II 809683 | 6316 6320
#> [471361] II 808609 --- II 809917 | 6316 6324
#> [471362] II 808617 --- II 809506 | 6316 6319
#> [471363] II 809447 --- II 809685 | 6319 6321
#> [471364] II 809472 --- II 809675 | 6319 6320
#> distance
#> <integer>
#> [1] 48443
#> [2] 44890
#> [3] 687132
#> [4] 25964
#> [5] 38883
#> ... ...
#> [471360] 1078
#> [471361] 1308
#> [471362] 889
#> [471363] 238
#> [471364] 203
#> -------
#> regions: 549331 ranges and 0 metadata columns
#> seqinfo: 17 sequences from an unspecified genome
## --- HicFile
hic_file <- HiContactsData('yeast_wt', format = 'hic')
#> see ?HiContactsData and browseVignettes('HiContactsData') for documentation
#> loading from cache
hicf <- HicFile(hic_file, pairsFile = pairs_file)
hicf
#> HicFile object
#> .hic file: /github/home/.cache/R/ExperimentHub/3e265ac673a5_7836
#> resolution: 1000
#> pairs file: /github/home/.cache/R/ExperimentHub/3e2648419160_7753
#> metadata(0):
import(hicf)
#> `HiCExperiment` object with 13,681,280 contacts over 12,165 regions
#> -------
#> fileName: "/github/home/.cache/R/ExperimentHub/3e265ac673a5_7836"
#> focus: "whole genome"
#> resolutions(5): 1000 2000 4000 8000 16000
#> active resolution: 1000
#> interactions: 2965693
#> scores(2): count balanced
#> topologicalFeatures: compartments(0) borders(0) loops(0) viewpoints(0)
#> pairsFile: /github/home/.cache/R/ExperimentHub/3e2648419160_7753
#> metadata(0):
## --- HicproFile
hicpro_matrix_file <- HiContactsData('yeast_wt', format = 'hicpro_matrix')
#> see ?HiContactsData and browseVignettes('HiContactsData') for documentation
#> loading from cache
hicpro_regions_file <- HiContactsData('yeast_wt', format = 'hicpro_bed')
#> see ?HiContactsData and browseVignettes('HiContactsData') for documentation
#> loading from cache
hicprof <- HicproFile(hicpro_matrix_file, bed = hicpro_regions_file)
hicprof
#> HicproFile object
#> HiC-Pro files:
#> $ matrix: /github/home/.cache/R/ExperimentHub/3e2639eaefe7_7837
#> $ regions: /github/home/.cache/R/ExperimentHub/3e2612bef73a_7838
#> resolution: 1000
#> pairs file:
#> metadata(0):
import(hicprof)
#> Registered S3 methods overwritten by 'readr':
#> method from
#> as.data.frame.spec_tbl_df vroom
#> as_tibble.spec_tbl_df vroom
#> format.col_spec vroom
#> print.col_spec vroom
#> print.collector vroom
#> print.date_names vroom
#> print.locale vroom
#> str.col_spec vroom
#> `HiCExperiment` object with 9,503,604 contacts over 12,165 regions
#> -------
#> fileName: "/github/home/.cache/R/ExperimentHub/3e2639eaefe7_7837"
#> focus: "whole genome"
#> resolutions(1): 1000
#> active resolution: 1000
#> interactions: 2686250
#> scores(2): count balanced
#> topologicalFeatures: compartments(0) borders(0) loops(0) viewpoints(0)
#> pairsFile: N/A
#> metadata(1): regionsImport arguments
Querying subsets of Hi-C matrix files
The focus argument is used to specifically import
contacts within a genomic locus of interest.
availableChromosomes(cool_file)
#> Seqinfo object with 16 sequences from an unspecified genome:
#> seqnames seqlengths isCircular genome
#> I 230218 <NA> <NA>
#> II 813184 <NA> <NA>
#> III 316620 <NA> <NA>
#> IV 1531933 <NA> <NA>
#> V 576874 <NA> <NA>
#> ... ... ... ...
#> XII 1078177 <NA> <NA>
#> XIII 924431 <NA> <NA>
#> XIV 784333 <NA> <NA>
#> XV 1091291 <NA> <NA>
#> XVI 948066 <NA> <NA>
hic <- import(cool_file, format = 'cool', focus = 'I:20001-80000')
hic
#> `HiCExperiment` object with 24,322 contacts over 60 regions
#> -------
#> fileName: "/github/home/.cache/R/ExperimentHub/3e2645368057_7751"
#> focus: "I:20,001-80,000"
#> resolutions(1): 1000
#> active resolution: 1000
#> interactions: 1653
#> scores(2): count balanced
#> topologicalFeatures: compartments(0) borders(0) loops(0) viewpoints(0)
#> pairsFile: N/A
#> metadata(0):
focus(hic)
#> [1] "I:20001-80000"Note:
Querying subsets of HiC-Pro formatted matrices is currently not
supported. HiC-Pro formatted matrices will systematically be fully
imported in memory when imported.
One can also extract a count matrix from a Hi-C matrix file that is
not centered at the diagonal. To do this, specify a couple of
coordinates in the focus argument using a character string
formatted as "...|...":
Multi-resolution Hi-C matrix files
import() works with .mcool and
multi-resolution .hic files as well: in this case, the user
can specify the resolution at which count values are
recovered.
mcool_file <- HiContactsData('yeast_wt', format = 'mcool')
#> see ?HiContactsData and browseVignettes('HiContactsData') for documentation
#> loading from cache
availableResolutions(mcool_file)
#> resolutions(5): 1000 2000 4000 8000 16000
#>
availableChromosomes(mcool_file)
#> Seqinfo object with 16 sequences from an unspecified genome:
#> seqnames seqlengths isCircular genome
#> I 230218 <NA> <NA>
#> II 813184 <NA> <NA>
#> III 316620 <NA> <NA>
#> IV 1531933 <NA> <NA>
#> V 576874 <NA> <NA>
#> ... ... ... ...
#> XII 1078177 <NA> <NA>
#> XIII 924431 <NA> <NA>
#> XIV 784333 <NA> <NA>
#> XV 1091291 <NA> <NA>
#> XVI 948066 <NA> <NA>
hic <- import(mcool_file, format = 'cool', focus = 'II:1-800000', resolution = 2000)
hic
#> `HiCExperiment` object with 466,123 contacts over 400 regions
#> -------
#> fileName: "/github/home/.cache/R/ExperimentHub/3e261f048867_7752"
#> focus: "II:1-800,000"
#> resolutions(5): 1000 2000 4000 8000 16000
#> active resolution: 2000
#> interactions: 33479
#> scores(2): count balanced
#> topologicalFeatures: compartments(0) borders(0) loops(0) viewpoints(0)
#> pairsFile: N/A
#> metadata(0):HiCExperiment accessors
Slots
Slots for a HiCExperiment object can be accessed using
the following getters:
fileName(hic)
#> [1] "/github/home/.cache/R/ExperimentHub/3e261f048867_7752"
focus(hic)
#> [1] "II:1-800000"
resolutions(hic)
#> [1] 1000 2000 4000 8000 16000
resolution(hic)
#> [1] 2000
interactions(hic)
#> GInteractions object with 33479 interactions and 4 metadata columns:
#> seqnames1 ranges1 seqnames2 ranges2 | bin_id1
#> <Rle> <IRanges> <Rle> <IRanges> | <numeric>
#> [1] II 1-2000 --- II 1-2000 | 116
#> [2] II 1-2000 --- II 4001-6000 | 116
#> [3] II 1-2000 --- II 6001-8000 | 116
#> [4] II 1-2000 --- II 8001-10000 | 116
#> [5] II 1-2000 --- II 10001-12000 | 116
#> ... ... ... ... ... ... . ...
#> [33475] II 794001-796000 --- II 796001-798000 | 513
#> [33476] II 794001-796000 --- II 798001-800000 | 513
#> [33477] II 796001-798000 --- II 796001-798000 | 514
#> [33478] II 796001-798000 --- II 798001-800000 | 514
#> [33479] II 798001-800000 --- II 798001-800000 | 515
#> bin_id2 count balanced
#> <numeric> <numeric> <numeric>
#> [1] 116 1 NaN
#> [2] 118 2 NaN
#> [3] 119 3 NaN
#> [4] 120 15 NaN
#> [5] 121 9 NaN
#> ... ... ... ...
#> [33475] 514 309 0.1194189
#> [33476] 515 227 0.0956207
#> [33477] 514 130 0.0501703
#> [33478] 515 297 0.1249314
#> [33479] 515 117 0.0536429
#> -------
#> regions: 400 ranges and 4 metadata columns
#> seqinfo: 16 sequences from an unspecified genome
scores(hic)
#> List of length 2
#> names(2): count balanced
tail(scores(hic, 1))
#> [1] 212 309 227 130 297 117
tail(scores(hic, 'balanced'))
#> [1] 0.08204677 0.11941893 0.09562069 0.05017035 0.12493137 0.05364290
topologicalFeatures(hic)
#> List of length 4
#> names(4): compartments borders loops viewpoints
pairsFile(hic)
#> NULL
metadata(hic)
#> list()Several extra functions are available as well:
seqinfo(hic) ## To recover the `Seqinfo` object from the `.(m)cool` file
#> Seqinfo object with 16 sequences from an unspecified genome:
#> seqnames seqlengths isCircular genome
#> I 230218 <NA> <NA>
#> II 813184 <NA> <NA>
#> III 316620 <NA> <NA>
#> IV 1531933 <NA> <NA>
#> V 576874 <NA> <NA>
#> ... ... ... ...
#> XII 1078177 <NA> <NA>
#> XIII 924431 <NA> <NA>
#> XIV 784333 <NA> <NA>
#> XV 1091291 <NA> <NA>
#> XVI 948066 <NA> <NA>
bins(hic) ## To bin the genome at the current resolution
#> GRanges object with 6045 ranges and 2 metadata columns:
#> seqnames ranges strand | bin_id weight
#> <Rle> <IRanges> <Rle> | <numeric> <numeric>
#> I_1_2000 I 1-2000 * | 0 0.0559613
#> I_2001_4000 I 2001-4000 * | 1 0.0333136
#> I_4001_6000 I 4001-6000 * | 2 0.0376028
#> I_6001_8000 I 6001-8000 * | 3 0.0369553
#> I_8001_10000 I 8001-10000 * | 4 0.0220139
#> ... ... ... ... . ... ...
#> XVI_940001_942000 XVI 940001-942000 * | 6040 0.0226033
#> XVI_942001_944000 XVI 942001-944000 * | 6041 NaN
#> XVI_944001_946000 XVI 944001-946000 * | 6042 NaN
#> XVI_946001_948000 XVI 946001-948000 * | 6043 NaN
#> XVI_948001_948066 XVI 948001-948066 * | 6044 NaN
#> -------
#> seqinfo: 16 sequences from an unspecified genome
regions(hic) ## To extract unique regions of the contact matrix
#> GRanges object with 400 ranges and 4 metadata columns:
#> seqnames ranges strand | bin_id weight chr
#> <Rle> <IRanges> <Rle> | <numeric> <numeric> <Rle>
#> II_1_2000 II 1-2000 * | 116 NaN II
#> II_2001_4000 II 2001-4000 * | 117 NaN II
#> II_4001_6000 II 4001-6000 * | 118 NaN II
#> II_6001_8000 II 6001-8000 * | 119 NaN II
#> II_8001_10000 II 8001-10000 * | 120 0.0461112 II
#> ... ... ... ... . ... ... ...
#> II_790001_792000 II 790001-792000 * | 511 0.0236816 II
#> II_792001_794000 II 792001-794000 * | 512 0.0272236 II
#> II_794001_796000 II 794001-796000 * | 513 0.0196726 II
#> II_796001_798000 II 796001-798000 * | 514 0.0196450 II
#> II_798001_800000 II 798001-800000 * | 515 0.0214123 II
#> center
#> <integer>
#> II_1_2000 1000
#> II_2001_4000 3000
#> II_4001_6000 5000
#> II_6001_8000 7000
#> II_8001_10000 9000
#> ... ...
#> II_790001_792000 791000
#> II_792001_794000 793000
#> II_794001_796000 795000
#> II_796001_798000 797000
#> II_798001_800000 799000
#> -------
#> seqinfo: 16 sequences from an unspecified genome
anchors(hic) ## To extract "first" and "second" anchors for each interaction
#> $first
#> GRanges object with 33479 ranges and 4 metadata columns:
#> seqnames ranges strand | bin_id weight chr center
#> <Rle> <IRanges> <Rle> | <numeric> <numeric> <Rle> <integer>
#> [1] II 1-2000 * | 116 NaN II 1000
#> [2] II 1-2000 * | 116 NaN II 1000
#> [3] II 1-2000 * | 116 NaN II 1000
#> [4] II 1-2000 * | 116 NaN II 1000
#> [5] II 1-2000 * | 116 NaN II 1000
#> ... ... ... ... . ... ... ... ...
#> [33475] II 794001-796000 * | 513 0.0196726 II 795000
#> [33476] II 794001-796000 * | 513 0.0196726 II 795000
#> [33477] II 796001-798000 * | 514 0.0196450 II 797000
#> [33478] II 796001-798000 * | 514 0.0196450 II 797000
#> [33479] II 798001-800000 * | 515 0.0214123 II 799000
#> -------
#> seqinfo: 16 sequences from an unspecified genome
#>
#> $second
#> GRanges object with 33479 ranges and 4 metadata columns:
#> seqnames ranges strand | bin_id weight chr center
#> <Rle> <IRanges> <Rle> | <numeric> <numeric> <Rle> <integer>
#> [1] II 1-2000 * | 116 NaN II 1000
#> [2] II 4001-6000 * | 118 NaN II 5000
#> [3] II 6001-8000 * | 119 NaN II 7000
#> [4] II 8001-10000 * | 120 0.0461112 II 9000
#> [5] II 10001-12000 * | 121 0.0334807 II 11000
#> ... ... ... ... . ... ... ... ...
#> [33475] II 796001-798000 * | 514 0.0196450 II 797000
#> [33476] II 798001-800000 * | 515 0.0214123 II 799000
#> [33477] II 796001-798000 * | 514 0.0196450 II 797000
#> [33478] II 798001-800000 * | 515 0.0214123 II 799000
#> [33479] II 798001-800000 * | 515 0.0214123 II 799000
#> -------
#> seqinfo: 16 sequences from an unspecified genomeSlot setters
Features
Add topologicalFeatures using GRanges or
Pairs.
topologicalFeatures(hic, 'viewpoints') <- GRanges("II:300001-320000")
topologicalFeatures(hic)
#> List of length 4
#> names(4): compartments borders loops viewpoints
topologicalFeatures(hic, 'viewpoints')
#> GRanges object with 1 range and 0 metadata columns:
#> seqnames ranges strand
#> <Rle> <IRanges> <Rle>
#> [1] II 300001-320000 *
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengthsCoercing HiCExperiment
Using the as() function, HiCExperiment can
be coerced in GInteractions, ContactMatrix and
matrix seamlessly.
as(hic, "GInteractions")
#> GInteractions object with 33479 interactions and 5 metadata columns:
#> seqnames1 ranges1 seqnames2 ranges2 | bin_id1
#> <Rle> <IRanges> <Rle> <IRanges> | <numeric>
#> [1] II 1-2000 --- II 1-2000 | 116
#> [2] II 1-2000 --- II 4001-6000 | 116
#> [3] II 1-2000 --- II 6001-8000 | 116
#> [4] II 1-2000 --- II 8001-10000 | 116
#> [5] II 1-2000 --- II 10001-12000 | 116
#> ... ... ... ... ... ... . ...
#> [33475] II 794001-796000 --- II 796001-798000 | 513
#> [33476] II 794001-796000 --- II 798001-800000 | 513
#> [33477] II 796001-798000 --- II 796001-798000 | 514
#> [33478] II 796001-798000 --- II 798001-800000 | 514
#> [33479] II 798001-800000 --- II 798001-800000 | 515
#> bin_id2 count balanced random
#> <numeric> <numeric> <numeric> <numeric>
#> [1] 116 1 NaN 0.08075014
#> [2] 118 2 NaN 0.83433304
#> [3] 119 3 NaN 0.60076089
#> [4] 120 15 NaN 0.15720844
#> [5] 121 9 NaN 0.00739944
#> ... ... ... ... ...
#> [33475] 514 309 0.1194189 0.704594
#> [33476] 515 227 0.0956207 0.274246
#> [33477] 514 130 0.0501703 0.961907
#> [33478] 515 297 0.1249314 0.825239
#> [33479] 515 117 0.0536429 0.664753
#> -------
#> regions: 400 ranges and 4 metadata columns
#> seqinfo: 16 sequences from an unspecified genome
as(hic, "ContactMatrix")
#> class: ContactMatrix
#> dim: 400 400
#> type: dgCMatrix
#> rownames: NULL
#> colnames: NULL
#> metadata(0):
#> regions: 400
as(hic, "matrix")[1:10, 1:10]
#> [,1] [,2] [,3] [,4] [,5] [,6] [,7] [,8]
#> [1,] NaN 0 NaN NaN NaN NaN NaN NaN
#> [2,] 0 0 0 0 0.00000000 0.00000000 0.00000000 0.00000000
#> [3,] NaN 0 0 NaN NaN NaN NaN NaN
#> [4,] NaN 0 NaN NaN NaN NaN NaN NaN
#> [5,] NaN 0 NaN NaN 0.08079721 0.18680431 0.13127403 0.08833001
#> [6,] NaN 0 NaN NaN 0.18680431 0.08183011 0.19176749 0.12687633
#> [7,] NaN 0 NaN NaN 0.13127403 0.19176749 0.08040523 0.13690173
#> [8,] NaN 0 NaN NaN 0.08833001 0.12687633 0.13690173 0.07977117
#> [9,] NaN 0 NaN NaN 0.06759757 0.10078115 0.13249106 0.18151495
#> [10,] NaN 0 NaN NaN 0.06021225 0.07728955 0.09404388 0.12720548
#> [,9] [,10]
#> [1,] NaN NaN
#> [2,] 0.00000000 0.00000000
#> [3,] NaN NaN
#> [4,] NaN NaN
#> [5,] 0.06759757 0.06021225
#> [6,] 0.10078115 0.07728955
#> [7,] 0.13249106 0.09404388
#> [8,] 0.18151495 0.12720548
#> [9,] 0.06494950 0.11622354
#> [10,] 0.11622354 0.06796588
as(hic, "data.frame")[1:10, ]
#> seqnames1 start1 end1 width1 strand1 bin_id1 weight1 center1 seqnames2
#> 1 II 1 2000 2000 * 116 NaN 1000 II
#> 2 II 1 2000 2000 * 116 NaN 1000 II
#> 3 II 1 2000 2000 * 116 NaN 1000 II
#> 4 II 1 2000 2000 * 116 NaN 1000 II
#> 5 II 1 2000 2000 * 116 NaN 1000 II
#> 6 II 1 2000 2000 * 116 NaN 1000 II
#> 7 II 1 2000 2000 * 116 NaN 1000 II
#> 8 II 1 2000 2000 * 116 NaN 1000 II
#> 9 II 1 2000 2000 * 116 NaN 1000 II
#> 10 II 1 2000 2000 * 116 NaN 1000 II
#> start2 end2 width2 strand2 bin_id2 weight2 center2 count balanced
#> 1 1 2000 2000 * 116 NaN 1000 1 NaN
#> 2 4001 6000 2000 * 118 NaN 5000 2 NaN
#> 3 6001 8000 2000 * 119 NaN 7000 3 NaN
#> 4 8001 10000 2000 * 120 0.04611120 9000 15 NaN
#> 5 10001 12000 2000 * 121 0.03348075 11000 9 NaN
#> 6 12001 14000 2000 * 122 0.03389168 13000 6 NaN
#> 7 14001 16000 2000 * 123 0.04164320 15000 1 NaN
#> 8 16001 18000 2000 * 124 0.01954625 17000 2 NaN
#> 9 18001 20000 2000 * 125 0.02331795 19000 6 NaN
#> 10 20001 22000 2000 * 126 0.02241734 21000 5 NaN
#> random
#> 1 0.080750138
#> 2 0.834333037
#> 3 0.600760886
#> 4 0.157208442
#> 5 0.007399441
#> 6 0.466393497
#> 7 0.497777389
#> 8 0.289767245
#> 9 0.732881987
#> 10 0.772521511Importing pairs files
Pairs files typically contain chimeric pairs (filtered after mapping), corresponding to loci that have been religated together after restriction enzyme digestion. Such files have a variety of standards.
- The
.pairsfile format, supported by the 4DN consortium:[ ] - The pairs format generated by Juicer: [
] [ ] [ ] [ ] - The
.(all)validPairsfile format, defined in the HiC-Pro pipeline:[ ]
Pairs in any of these different formats are automatically detected
and imported in R with the import function:
import(pairs_file, format = 'pairs')
#> GInteractions object with 471364 interactions and 3 metadata columns:
#> seqnames1 ranges1 seqnames2 ranges2 | frag1 frag2
#> <Rle> <IRanges> <Rle> <IRanges> | <numeric> <numeric>
#> [1] II 105 --- II 48548 | 1358 1681
#> [2] II 113 --- II 45003 | 1358 1658
#> [3] II 119 --- II 687251 | 1358 5550
#> [4] II 160 --- II 26124 | 1358 1510
#> [5] II 169 --- II 39052 | 1358 1613
#> ... ... ... ... ... ... . ... ...
#> [471360] II 808605 --- II 809683 | 6316 6320
#> [471361] II 808609 --- II 809917 | 6316 6324
#> [471362] II 808617 --- II 809506 | 6316 6319
#> [471363] II 809447 --- II 809685 | 6319 6321
#> [471364] II 809472 --- II 809675 | 6319 6320
#> distance
#> <integer>
#> [1] 48443
#> [2] 44890
#> [3] 687132
#> [4] 25964
#> [5] 38883
#> ... ...
#> [471360] 1078
#> [471361] 1308
#> [471362] 889
#> [471363] 238
#> [471364] 203
#> -------
#> regions: 549331 ranges and 0 metadata columns
#> seqinfo: 17 sequences from an unspecified genomeFurther documentation
Please check ?HiCExperiment in R for a full description
of available slots, getters and setters, and comprehensive examples of
interaction with a HiCExperiment object.
Session info
sessionInfo()
#> R version 4.6.0 (2026-04-24)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
#> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats4 stats graphics grDevices utils datasets methods
#> [8] base
#>
#> other attached packages:
#> [1] HiCExperiment_1.11.1 HiContactsData_1.13.0 ExperimentHub_3.2.0
#> [4] AnnotationHub_4.2.0 BiocFileCache_3.2.0 dbplyr_2.5.2
#> [7] GenomicRanges_1.64.0 Seqinfo_1.2.0 IRanges_2.46.0
#> [10] S4Vectors_0.50.0 BiocGenerics_0.58.0 generics_0.1.4
#> [13] dplyr_1.2.1 BiocStyle_2.40.0
#>
#> loaded via a namespace (and not attached):
#> [1] DBI_1.3.0 httr2_1.2.2
#> [3] rlang_1.2.0 magrittr_2.0.5
#> [5] otel_0.2.0 matrixStats_1.5.0
#> [7] compiler_4.6.0 RSQLite_2.4.6
#> [9] png_0.1-9 systemfonts_1.3.2
#> [11] vctrs_0.7.3 stringr_1.6.0
#> [13] pkgconfig_2.0.3 crayon_1.5.3
#> [15] fastmap_1.2.0 XVector_0.52.0
#> [17] rmarkdown_2.31 tzdb_0.5.0
#> [19] ggbeeswarm_0.7.3 strawr_0.0.92
#> [21] ragg_1.5.2 purrr_1.2.2
#> [23] bit_4.6.0 xfun_0.57
#> [25] cachem_1.1.0 jsonlite_2.0.0
#> [27] blob_1.3.0 rhdf5filters_1.24.0
#> [29] DelayedArray_0.38.0 Rhdf5lib_2.0.0
#> [31] BiocParallel_1.45.0 parallel_4.6.0
#> [33] R6_2.6.1 bslib_0.10.0
#> [35] stringi_1.8.7 RColorBrewer_1.1-3
#> [37] jquerylib_0.1.4 Rcpp_1.1.1-1.1
#> [39] bookdown_0.46 SummarizedExperiment_1.42.0
#> [41] knitr_1.51 readr_2.2.0
#> [43] Matrix_1.7-5 tidyselect_1.2.1
#> [45] abind_1.4-8 yaml_2.3.12
#> [47] codetools_0.2-20 curl_7.1.0
#> [49] lattice_0.22-9 tibble_3.3.1
#> [51] InteractionSet_1.40.0 Biobase_2.72.0
#> [53] withr_3.0.2 KEGGREST_1.52.0
#> [55] S7_0.2.2 evaluate_1.0.5
#> [57] ggrastr_1.0.2 desc_1.4.3
#> [59] Biostrings_2.80.0 pillar_1.11.1
#> [61] BiocManager_1.30.27 filelock_1.0.3
#> [63] MatrixGenerics_1.24.0 vroom_1.7.1
#> [65] hms_1.1.4 BiocVersion_3.23.1
#> [67] ggplot2_4.0.3 scales_1.4.0
#> [69] glue_1.8.1 tools_4.6.0
#> [71] BiocIO_1.22.0 RSpectra_0.16-2
#> [73] fs_2.1.0 rhdf5_2.56.0
#> [75] grid_4.6.0 tidyr_1.3.2
#> [77] AnnotationDbi_1.74.0 beeswarm_0.4.0
#> [79] vipor_0.4.7 cli_3.6.6
#> [81] rappdirs_0.3.4 textshaping_1.0.5
#> [83] S4Arrays_1.12.0 gtable_0.3.6
#> [85] HiContacts_1.14.0 sass_0.4.10
#> [87] digest_0.6.39 SparseArray_1.12.0
#> [89] htmlwidgets_1.6.4 farver_2.1.2
#> [91] memoise_2.0.1 htmltools_0.5.9
#> [93] pkgdown_2.2.0 lifecycle_1.0.5
#> [95] httr_1.4.8 bit64_4.8.0